18017847121

《Advanced science》是Wiley出版集团旗下的一本开放获取(Open Access)综合性学术期刊,于2014年创刊。在中科院最新升级版分区表中,大类学科为材料科学1区,小类学科中化学综合为1区、纳米科技为2区、材料科学综合为1区,属于TOP期刊。《Advanced science》是一本跨学科开放获取期刊,涵盖材料科学、物理和化学、医学和生命科学以及工程学等领域,主要发表这些领域的yi流基础和应用研究成果。
出版周期:12 issues/year;
影响因子:14.1;
ISSN:2198-3844;
发文量:3290篇/年;
审稿速度:平均12周;
版面费:USD 5270.00;

一、研究背景与目标
肺纤维化(PF)是多种肺部疾病的进展性致命结局,特发性肺纤维化(IPF)为主要亚型,由慢性肺部炎症驱动,患者确诊后平均生存期仅3-5年。全球发病率上升,新冠疫情后PF发生率达2-6%,约44.9%的新冠幸存者发展为PF。临床仅吡非尼酮和尼达尼布获批,疗效有限,亟需阐明发病机制以开发新疗法。成纤维细胞胶原合成是PF的核心特征,但其异常激活的触发因素不明。本研究聚焦组织蛋白酶K(CTSK)在PF中的作用及机制。
二、关键技术总结
本文为探究组织蛋白酶 K(CTSK)在肺纤维化(PF)中的作用及机制,采用了多种实验技术,涵盖分子生物学、细胞生物学、动物模型及临床样本分析等领域,具体如下:
1. 肺纤维化模型:通过气管内注射博来霉素(BLM)构建小鼠PF模型,用于评估CTSK对纤维化进展的影响;通过百cao枯(PQ)诱导急性肺损伤(ALI)模型,结合不同时间点采样进行蛋白质组学分析。
2. 基因编辑模型:构建成纤维细胞特异性CTSK条件敲除小鼠(Col1a2-CreERT;CTSK flox/flox),验证内源性CTSK的作用。
3. 药物干预:对模型小鼠施用重组CTSK(rCTSK)、CTSK抑制剂(odanacatib)、内吞抑制剂(Pitstop2)、SMAD3抑制剂(SIS3)及谷氨酰胺酶抑制剂(DON等),探究相关通路的作用。
4. 蛋白质组学与单细胞测序(scRNA-seq):通过蛋白质组学筛选PF模型小鼠肺组织中差异表达的组织蛋白酶家族成员,结合scRNA-seq(数据集 GSE111664、GSE136831 等)定位CTSK在成纤维细胞中的富集。
5. 细胞模型:使用人成纤维细胞系(MRC-5),经TGF-β1诱导模拟成纤维细胞-肌成纤维细胞转化(FMT),研究CTSK对胶原合成的影响;分离原代小鼠肺成纤维细胞进行验证。
6. 蛋白相互作用验证:采用免疫共沉淀(Co-IP)验证CTSK与SNX9的直接相互作用;通过AlphaFold3预测结合位点,结合丙氨酸突变实验确定关键氨基酸残基。
7. 分子表达检测:利用Western blot检测蛋白质表达水平,实时定量PCR(qRT-PCR)分析 mRNA 水平,免疫荧光染色观察蛋白定位与共定位。
8. 非靶向与靶向代谢组学:通过LC-MS/MS对TGF-β1诱导的MRC-5细胞(经rCTSK处理)进行非靶向代谢组学分析,结合KEGG和MSEA富集分析筛选差异代谢通路(如谷氨酰胺代谢);通过靶向代谢组学检测谷氨酰胺、谷氨酸及脯氨酸等代谢物水平。
9. 组织与病理分析:采用Masson三色染色和Sirius Red染色检测肺组织胶原沉积;免疫组化分析CTSK、SNX9等蛋白在肺组织中的表达与定位;原子力显微镜检测肺组织硬度,评估纤维化程度。
10. 临床样本分析:收集PF患者及健康对照的肺组织、支气管肺泡灌洗液(BALF)和血清样本,通过ELISA测定血清和BALF中CTSK及谷氨酰胺水平;结合生存分析(Kaplan-Meier)评估CTSK与患者预后的相关性。
11. 转录组数据分析:利用公开RNA-seq数据集(GSE124685、GSE70866)分析CTSK家族成员在PF患者中的表达差异及与生存的关联。
三、主要研究结果
1、CTSK在PF进展过程中的积累
通过蛋白质组学分析PQ诱导ALI小鼠模型的肺组织发现,包括CTSK在内的组织蛋白酶家族成员(如CTSA、CTSB等)在ALI进展过程中(第1、3、7天)逐渐上调,其中CTSK 的积累最为显著。而在博来霉素(BLM)诱导的PF小鼠肺组织中,CTSK的mRNA水平较对照组显著升高,且通过免疫组化和Western blot在蛋白水平得到验证。同时,CTSK的表达在BLM诱导后逐渐增加,于第21天达到峰值,与模型中严重PF的发生时间一致。另外,基于BLM诱导PF小鼠的单细胞测序数据(GSE111664),CTSK在巨噬细胞和成纤维细胞中特异性上调,且成纤维细胞数量显著增加。此外, PF患者的单细胞测序数据(GSE136831)显示,成纤维细胞中CTSK也呈高表达;在经TGF-β1处理以模拟成纤维细胞-肌成纤维细胞转化(FMT)的人成纤维细胞系(MRC-5)中,CTSK在mRNA和蛋白水平均显著上调。

图1、CTSK在严重肺损伤及PF进展过程中在成纤维细胞中富集
2、CTSK对成纤维细胞胶原蛋白合成及肺纤维化PF进程的影响
对PF小鼠的单细胞测序数据(GSE132771)分析显示,CTSK阳性成纤维细胞中胶原蛋白相关基因(如 Col1a1、Col1a2、Col3a1)的表达显著高于CTSK阴性成纤维细胞,提示CTSK可能促进胶原蛋白合成。同时,在TGF-β1诱导的人成纤维细胞系(MRC-5)中,高浓度(0.1 μg/mL)的重组CTSK(rCTSK)经24小时处理后,可特异性上调COL1A1的表达,而对 COL1A2、COL3A1及α-SMA的表达无显著影响,且该结果在原代小鼠肺成纤维细胞中得到验证。另外,通过蛋白酶体抑制剂(MG132)和蛋白质合成抑制剂(环己酰亚胺)实验发现,rCTSK通过促进COL1A1的合成(而非抑制其降解)来增加其蛋白水平,且对α-SMA的表达无影响。在BLM诱导的PF小鼠模型中,施用rCTSK后,肺组织中COL1A1的表达和羟脯氨酸含量显著增加,Masson三色染色和Sirius Red染色显示胶原沉积和胶原纤维增多,原子力显微镜(AFM)证实肺组织硬度增加;同时,纤维化标志物mRNA水平也显著升高。rCTSK对纤维化进程的时序影响研究结果显示,BLM诱导后,在第7天施用rCTSK可显著上调第14天和第21天肺组织中CTSK和Col1a1的蛋白表达,且这些时间点的血清 CTSK水平也明显升高,表明过量CTSK在肺损伤后会加剧PF进展。综上,图2的结果证实CTSK可通过促进成纤维细胞中胶原蛋白(尤其是COL1A1)的合成,加剧肺纤维化的严重程度。

图2、CTSK可通过促进成纤维细胞中胶原蛋白的合成加剧肺纤维化的严重程度
3、CTSK与SNX9的相互作用及其对成纤维细胞胶原蛋白合成的调控机制
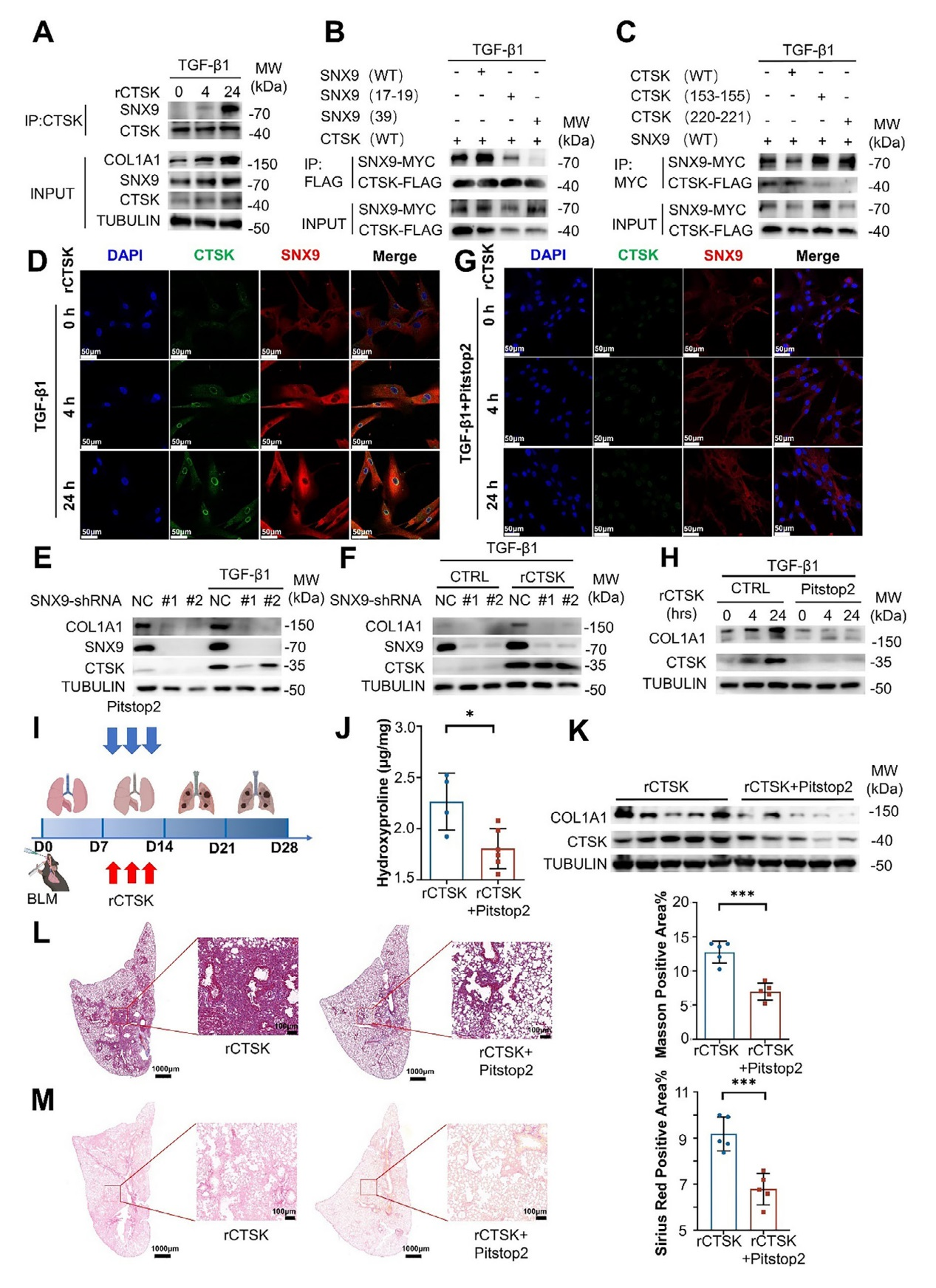
图3、CTSK与SNX9结合促进成纤维细胞中胶原蛋白合成进而加剧肺纤维化进程
通过免疫共沉淀(Co-IP)实验验证,在经TGF-β1和重组CTSK(rCTSK)处理的人成纤维细胞系(MRC-5)中,CTSK与SNX9存在直接相互作用,且随着处理时间延长,COL1A1 和SNX9的积累增加。AlphaFold3预测结合界面显示,CTSK的LYS220/CYS221残基与 SNX9的TRP39残基是相互作用的关键位点,丙氨酸突变实验进一步证实了这些位点的重要性;免疫荧光显示CTSK与SNX9主要在细胞质和核膜共定位,提示CTSK可能通过与 SNX9结合被内吞。在MRC-5细胞中敲低SNX9后,TGF-β1诱导的COL1A1表达降低,且 rCTSK无法再上调COL1A1的产生,表明SNX9是CTSK促进胶原蛋白合成的关键介质。另外,使用网格蛋白介导的内吞作用(CME)抑制剂Pitstop2处理MRC-5细胞后,CTSK的细胞内积累减少,同时COL1A1的表达也显著降低,证实内吞过程在CTSK调控胶原蛋白合成中不ke缺少。此外,体内验证实验表面,在BLM诱导的肺纤维化小鼠模型中,联合施用rCTSK和Pitstop2后,与单独施用rCTSK的小鼠相比,肺组织中羟脯氨酸含量、COL1A1 和CTSK蛋白水平显著降低,Masson和Sirius Red染色显示胶原沉积减少,纤维化标志物的mRNA水平也下降,表明阻断SNX9介导的内吞可缓解CTSK加剧的肺纤维化。该部分结果揭示CTSK通过与SNX9结合并经内吞作用,促进成纤维细胞中胶原蛋白合成,进而加剧肺纤维化进程。
4、CTSK通过TGF-β1-SMAD3信号通路促进胶原蛋白合成的机制研究
在经TGF-β1和重组CTSK(rCTSK)处理的人成纤维细胞系(MRC-5)中,SMAD3的磷酸化水平随处理时间延长显著升高,且与COL1A1的表达增加同步;在BLM诱导的肺纤维化小鼠模型中,施用rCTSK后,肺组织中磷酸化SMAD3(p-SMAD3)、COL1A1、SNX9和 CTSK的蛋白水平均上调。而在MRC-5细胞中敲低SNX9后,rCTSK增强的胞质p-SMAD3水平显著降低,且核内p-SMAD3的积累也大幅减少;免疫荧光实验进一步证实,SNX9敲低后,p-SMAD3与SNX9的共定位减少,表明SNX9是CTSK激活SMAD3的关键介质。免疫共沉淀(Co-IP)实验显示,CTSK、SNX9与p-SMAD3之间存在关联,提示三者可能形成复合物共同参与信号传递。此外,使用SMAD3抑制剂SIS3处理MRC-5细胞后,无论是基础状态还是rCTSK诱导的COL1A1表达均被显著抑制,同时细胞内CTSK水平也降低,证实SMAD3激活是CTSK促进胶原蛋白合成的必要环节。图4的结果揭示CTSK通过 SNX9介导的内吞作用,激活TGF-β1诱导的SMAD3磷酸化,进而促进成纤维细胞中胶原蛋白的合成。

图4、CTSK通过SNX9介导的内吞作用,激活TGF-β1诱导的SMAD3磷酸化,进而促进成纤维细胞中胶原蛋白的合成
5、CTSK对谷氨酰胺代谢的调控及其在胶原蛋白合成中的作用
对PF患者的单细胞测序数据(GSE136831)分析显示,CTSK阳性成纤维细胞中谷氨酸显著富集,提示CTSK可能与谷氨酰胺代谢相关。同时,对经TGF-β1和重组CTSK(rCTSK)处理的MRC-5细胞进行非靶向代谢组学分析,KEGG和MSEA富集分析表明,rCTSK显著激活包括谷氨酰胺代谢在内的氨基酸相关通路,其中谷氨酰胺代谢通路因参与胶原蛋白合成所需的脯氨酸和甘氨酸生成而成为关键通路。靶向代谢组学检测显示,rCTSK处理后,TGF-β1诱导的MRC-5细胞中谷氨酰胺、谷氨酸和脯氨酸水平显著升高,其中谷氨酰胺和脯氨酸的增加更为明显。谷氨酰胺酶1(GLS1)是谷氨酰胺代谢的关键酶,rCTSK可上调TGF-β1诱导的MRC-5细胞中GLS1的mRNA和蛋白表达及酶活性;而敲低SNX9后,rCTSK对GLS1的上调作用被显著抑制,谷氨酰胺水平也随之降低,表明CTSK通过SNX9促进GLS1介导的谷氨酰胺代谢。综上,图5的结果揭示CTSK通过SNX9增强GLS1的表达和活性,激活谷氨酰胺代谢通路,为胶原蛋白合成提供原料,进而促进肺纤维化进程。
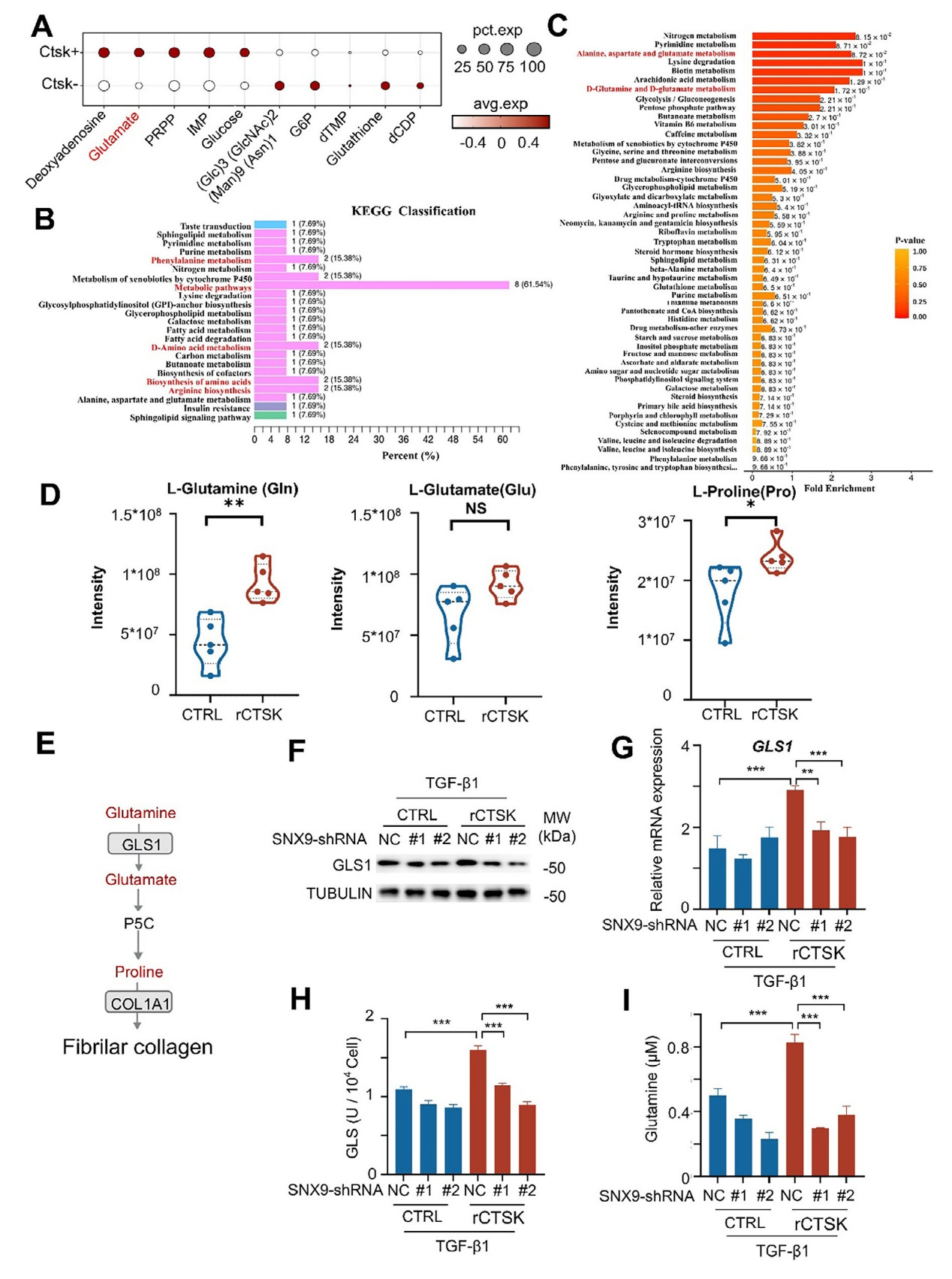
图5、CTSK高表达促进谷氨酰胺代谢
6、CTSK通过谷氨酰胺代谢促进成纤维细胞胶原蛋白合成的机制研究
在不含谷氨酰胺的培养基中,经TGF-β1和重组CTSK(rCTSK)处理的人成纤维细胞系(MRC-5)中,COL1A1的表达显著降低,而α-SMA水平不受影响;反之,在培养基中额外添加谷氨酰胺可显著增加COL1A1的产生,表明谷氨酰胺是CTSK促进胶原蛋白合成的关键原料。

图6、CTSK触发的谷氨酰胺代谢促进成纤维细胞胶原合成
另外,使用谷氨酰胺酶(GLS1)抑制剂(CB839、BPTES、DON)处理MRC-5细胞后,rCTSK 诱导的COL1A1表达被显著抑制,但α-SMA水平未受影响,证实GLS1介导的谷氨酰胺代谢在CTSK促进胶原蛋白合成中起核心作用。而在BLM诱导的肺纤维化小鼠模型中,联合施用rCTSK和DON(GLS1 抑制剂)后,与单独施用rCTSK的小鼠相比,肺组织中COL1A1的表达、羟脯氨酸含量显著降低,Masson三色染色和Sirius Red染色显示胶原沉积减少,纤维化标志物的mRNA水平也下降,表明抑制谷氨酰胺代谢可缓解CTSK加剧的肺纤维化。该部分的结果证实CTSK通过激活谷氨酰胺代谢通路促进成纤维细胞胶原蛋白合成,而抑制该代谢通路可有效减轻肺纤维化程度。
7、CTSK表达与谷氨酰胺水平及PF患者的预后相关性分析
对PF患者的RNA-seq数据(GSE124685)分析显示,包括CTSK在内的组织蛋白酶家族成员在肺组织中显著上调,其中CTSK的表达水平最高。免疫组化和Western blot验证显示,PF患者肺组织中CTSK、SNX9、GLS1及磷酸化SMAD3(p-SMAD3)的表达均显著高于健康对照;多重免疫荧光进一步证实这些分子在纤维化肺组织中存在共定位,提示它们在PF 发病中可能协同作用。此外,基于PF患者的RNA-seq数据(GSE70866)的生存分析显示,CTSK高表达患者的生存率显著低于低表达患者,表明CTSK可作为PF预后的潜在标志物。另外,PF患者的支气管肺泡灌洗液(BALF)和血清中CTSK水平显著高于健康对照及非 PF的急性呼吸窘迫综合征(ARDS)患者;且BALF和血清中CTSK水平高的患者死亡率显著升高。同时,PF患者血清中谷氨酰胺水平显著升高,且与CTSK水平呈正相关,进一步支持CTSK通过调控谷氨酰胺代谢影响PF进展的机制在临床中的相关性。结果表明,CTSK及其相关分子在PF患者中异常上调,血清CTSK水平可作为 PF 预后评估的潜在标志物,且其与谷氨酰胺水平的相关性为临床干预提供了方向。
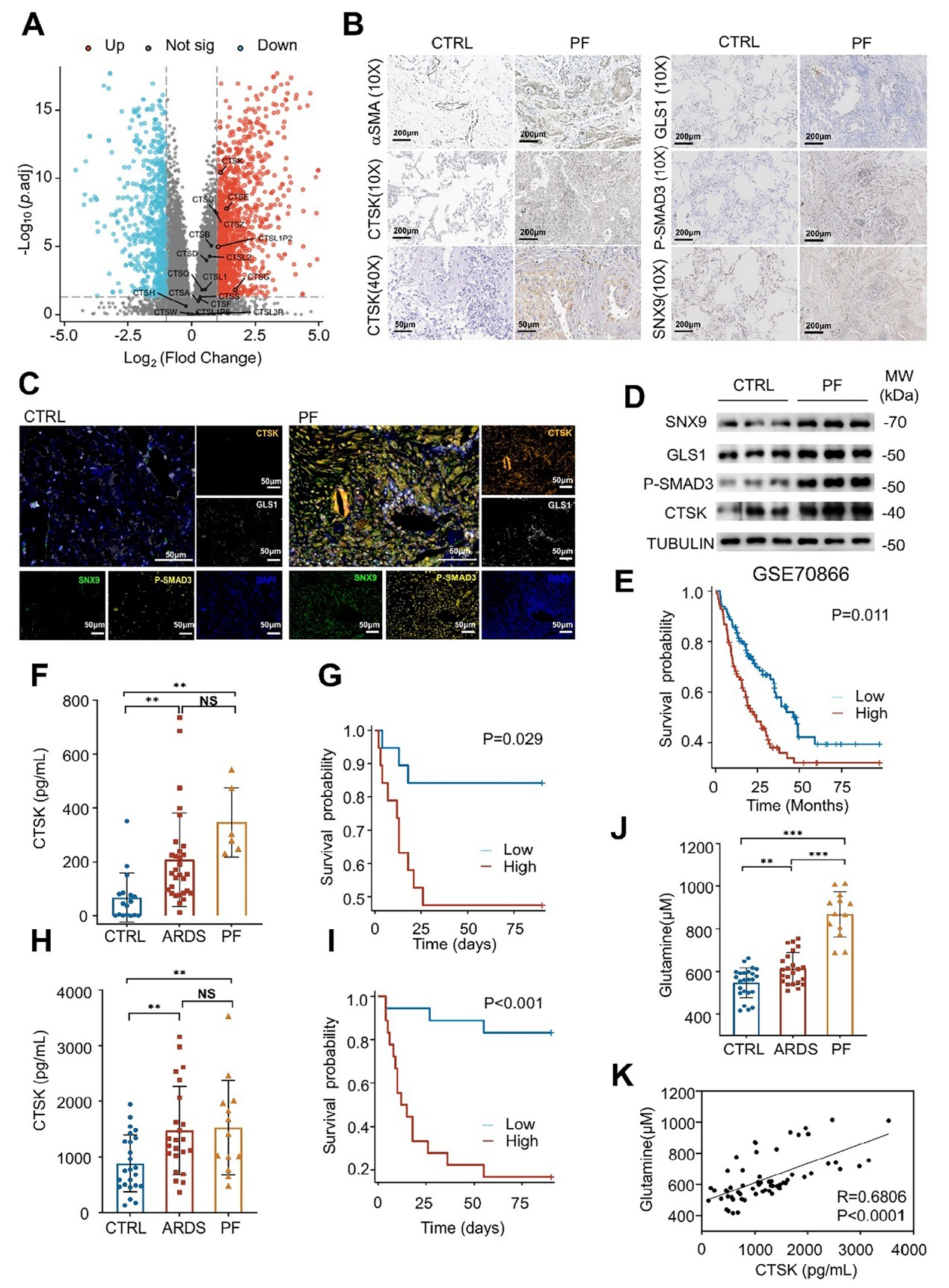
图7、患有肺纤维化(PF)的急性呼吸窘迫综合征(ARDS)患者中,外周组织蛋白酶 K(CTSK)含量与谷氨酰胺水平相关,且提示预后不良
四、全文结论
研究表明,组织蛋白酶 K(CTSK)在肺纤维化(PF)中通过双重机制发挥作用:早期降解细胞外基质,后期过量积累则与分选连接蛋白 9(SNX9)结合被内吞,激活TGF-β1-SMAD3 通路,上调谷氨酰胺酶 1(GLS1),促进谷氨酰胺代谢和胶原蛋白合成,加剧PF。临床数据显示,PF患者血清CTSK水平与谷氨酰胺水平正相关,且高CTSK预示不良预后。综上,CTSK和谷氨酰胺可作为PF预后标志物及治疗靶点。
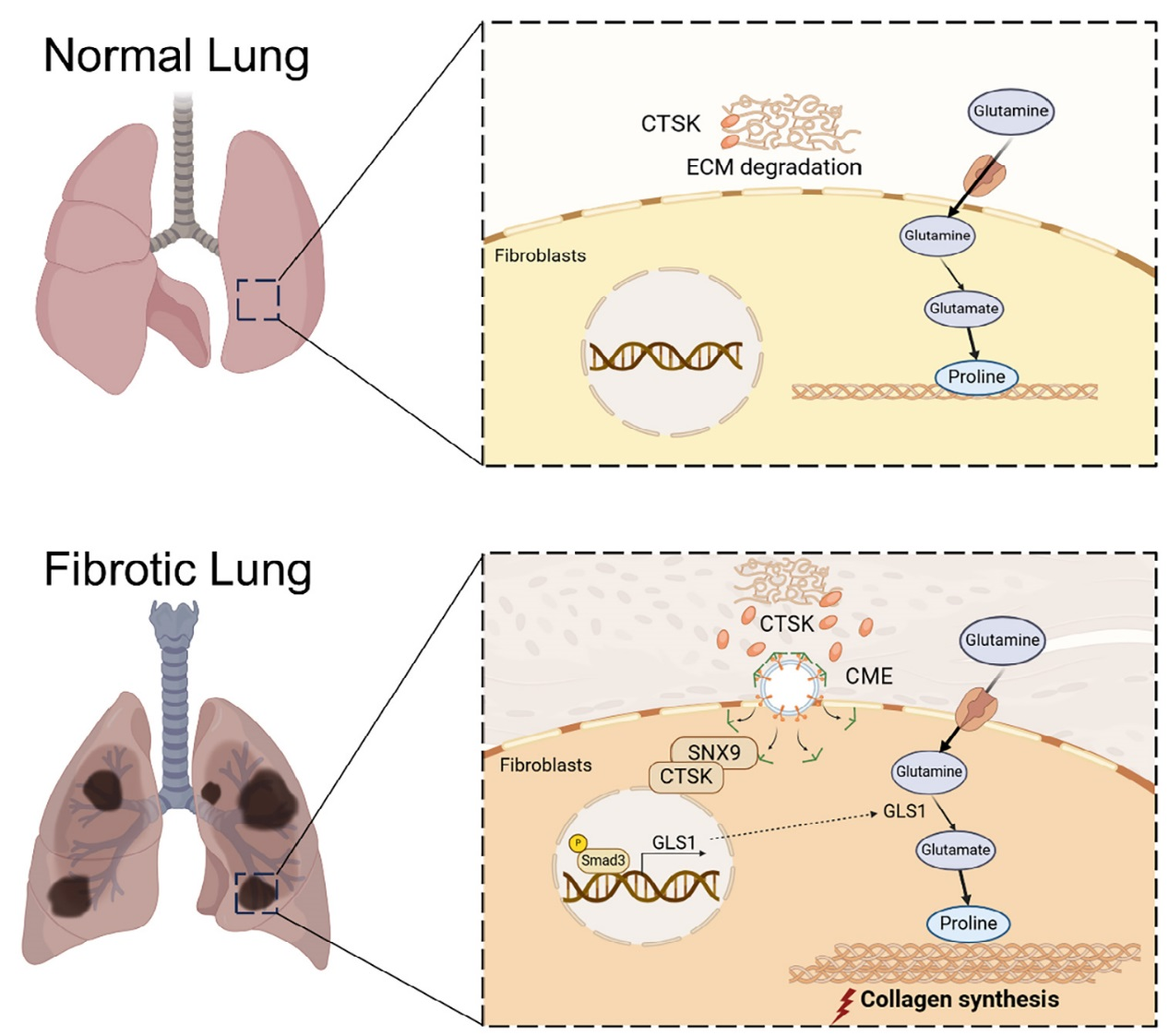
图8、流程图
参考文献:
Chen M, Meng X, Zhu Y, Wang D, Wang M, Wang Z, Tian X, Zhang J, Yue Z, Yang Z, Wang R. Cathepsin K Aggravates Pulmonary Fibrosis Through Promoting Fibroblast Glutamine Metabolism and Collagen Synthesis. Adv Sci (Weinh). 2025 Jul 3:e13017. doi: 10.1002/advs.202413017. Epub ahead of print. PMID: 40605618.
 欢迎来到
欢迎来到 联系人:张
联系人:张 地址:上海市长江南路180号长江软件园B区B637室
地址:上海市长江南路180号长江软件园B区B637室 邮箱:2844970554@qq.com
邮箱:2844970554@qq.com 传真:
传真: