18017847121

AR抑制剂不能增强PARP抑制剂对HR正常前列腺癌的疗效,反而通过阻滞细胞周期削弱其作用。DNA修复不受AR直接调控,协同作用缺乏理论支持。成果发表在PNAS杂志(IF:9.1);

《Proceedings of the National Academy of Sciences of the United States of America》简称PNAS,创刊于1915年,是美国国家科学院的guan方学术期刊,是shi界gong认的*综合性科学周刊之一。它致力于发表具有杰出原创性、重要科学贡献和广泛跨学科影响的前沿研究。发表涵盖生物科学、物理科学、社会科学、数学和工程学等几乎所有自然科学与社会科学分支的研究。年文章数约3000篇;提供混合开放获取模式。
当前,针对同源重组修复功能正常的去势抵抗性前列腺癌,临床前研究曾提出雄激素受体(AR)通路抑制剂与聚腺苷二磷酸核糖聚合酶(PARP)抑制剂联合治疗的协同作用假说。该假说基于AR可调控DNA损伤修复基因(如BRCA1/2)表达的理论,认为抑制AR通路可诱导“BRCAness"表型,从而增强肿瘤对PARP抑制剂的敏感性。然而,多项临床研究显示,该联合方案在HR正常患者中仅带来有限的生存获益,且美国食品药品监督管理局未批准其用于HR正常人群,凸显了现有理论与临床结果之间的显著矛盾。这一争议促使研究人员重新审视AR与DNA修复之间的调控关系,并深入探究在HR正常前列腺癌模型中AR抑制剂与PARP抑制剂相互作用的真实机制。
研究方法:
研究采用多种同源重组修复功能正常(HR-proficient)的前列腺癌细胞系(包括去势敏感型LNCaP和去势抵抗型22RV1、LNCaP-abl等)以及HR缺陷型对照细胞(BRCA2缺失的LuCaP176细胞),系统评估了雄激素受体靶向疗法与PARP抑制剂的相互作用。在方法学上,研究通过细胞活力检测(Cell Titer Glo)评估药物单用及联用的效果;利用γH2AX和RAD51免疫荧光染色定量分析DNA损伤与修复状态;采用RNA测序和ATAC测序分别在转录组和染色质可及性层面解析药物处理的分子变化;并通过免疫印迹验证关键蛋白表达。为验证细胞周期的作用,研究使用了CDK4/6抑制剂帕博西尼(palbociclib)和CDK7抑制剂SY-1365进行功能干预。此外,研究还结合了公共临床转录组数据,分析雄激素剥夺治疗(ADT)前后患者肿瘤中细胞周期相关通路的变化,从而在临床样本层面佐证机制发现。整体实验设计涵盖了从体外模型到临床关联的多层次验证,系统阐明了药物响应机制。
1. AR抑制在前列腺癌去势敏感或去势抵抗模型中均不与PARP抑制产生协同作用
在雄激素剥夺或使用恩杂鲁胺抑制AR信号后,去势敏感型前列腺癌细胞(LNCaP)对多种PARP抑制剂(包括奥拉帕利、卢卡帕利、他拉唑帕利及AZD-5305)的敏感性显著下降,表现为IC50值上升。然而,在去势抵抗型前列腺癌细胞系(22RV1、LN95、LNCaP-abl)中,AR抑制并未改变其对PARP抑制剂的敏感性。即使在临床相关浓度下(10 μM恩杂鲁胺与1 μM PARPi),联合用药也未能比单一用药显示出额外的细胞生长抑制优势。这些结果证明,AR抑制不仅无法与PARP抑制剂在HR正常的癌细胞中产生协同作用,反而在去势敏感模型中会削弱PARP抑制剂的疗效。

图1,AR抑制不改变去势抵抗性前列腺癌对PARP抑制的敏感性
2. PARP抑制剂治疗可调控AR相关基因的表达
随后,研究人员通过转录组分析揭示了PARP抑制剂在有无AR抑制条件下对HR正常前列腺癌细胞基因表达的影响。结果显示,PARP抑制剂处理显著诱导了CYP家族基因(如CYP1A、CYP1B)的表达。通路富集分析表明,在雄激素(DHT)存在的条件下,PARP抑制剂激活了p53通路并诱导细胞凋亡,同时抑制了细胞周期相关通路(如E2F靶标、G2/M检查点);而在雄激素剥夺条件下,这些细胞周期和凋亡通路的改变显著减弱甚至消失。这一发现与图1中观察到的生长抑制结果一致,表明PARP抑制剂的抗癌效果依赖于雄激素存在下的细胞周期进程,而非通过AR直接调控的DNA修复通路。

图2,PARP抑制诱导细胞周期和DNA损伤应答通路的变化
进一步的研究结果揭示了PARP抑制剂对AR驱动转录的调控作用及机制。RNA-seq分析显示,在DHT刺激的22RV1细胞中,PARP抑制剂能够抑制一部分雄激素响应基因的诱导表达(Cluster 2与Cluster 4),这些基因富集于细胞周期与代谢通路;而在LNCaP细胞中,PARP抑制剂仅对少量AR调控基因有抑制作用。ATAC-seq分析进一步发现,PARP抑制剂(如奥拉帕利)在DHT存在时会导致一部分染色质区域的可及性降低,但这种变化与基因表达改变的重合度很低。例如,LRRC4基因的可及性虽受DHT调控,却不受奥拉帕利影响。这些结果综合表明,PARP抑制剂对AR调控基因表达的影响主要在转录水平发生,而非通过改变染色质可及性来实现。

图3,PARP抑制剂对AR驱动转录的调控作用及机制
3. 雄激素剥夺或AR抑制不会抑制DNA修复。
RNA-seq分析显示,无论是在雄激素剥夺还是DHT处理的条件下,DNA修复相关基因(包括先前报道的AR靶基因如BRCA2)的表达均未受到显著抑制。ATAC-seq数据进一步证实,AR抑制并未改变BRCA1/BRCA2等关键修复基因位点的染色质可及性。功能实验通过γH2AX焦点形成检测发现,在去势敏感LNCaP细胞中,雄激素剥夺反而减弱了PARP抑制剂(奥拉帕利)诱导的DNA损伤;而在去势抵抗22RV1细胞中,DNA损伤水平不受雄激素状态影响。这些证据一致表明,AR通路抑制并不直接削弱前列腺癌细胞的DNA修复能力,也无法增强PARP抑制剂所致的DNA损伤。

图4,AR不直接调控DNA修复基因的表达
4. 细胞周期进展是响应PARP抑制的必要条件
研究发现,在去势敏感LNCaP细胞中,高剂量DHT(10 nM)诱导的细胞生长抑制会严重削弱奥拉帕利的疗效,其效果甚至强于雄激素剥夺。为验证这一现象是否存在于患者体内,研究人员分析了公共临床测序数据,发现雄激素剥夺治疗(ADT)显著抑制了患者肿瘤中细胞周期相关通路(E2F靶点、G2/M检查点)的基因表达,确认ADT在临床上确实会引起细胞周期停滞。为直接验证细胞周期的作用,研究使用CDK4/6抑制剂帕博西尼(Palbociclib)诱导G1期阻滞,发现这显著降低了多种PARP抑制剂(奥拉帕利、卢卡帕利、他拉唑帕利、AZD-5305)在HR正常前列腺癌细胞中的疗效。这种效应并不仅限于PARP抑制剂,同样也削弱了传统DNA损伤化疗药物(如顺铂、依托泊苷、多柔比星)的作用,表明细胞周期依赖性是DNA损伤类药物的普遍机制。有趣的是,在HR缺陷(BRCA2缺失)的LuCaP176细胞模型中,使用CDK7抑制剂SY-1365抑制细胞增殖,并未削弱奥拉帕利的疗效。这揭示了一个关键差异:细胞周期进展仅是HR正常细胞响应PARP抑制的必要条件,而在HR缺陷细胞中并非必需。
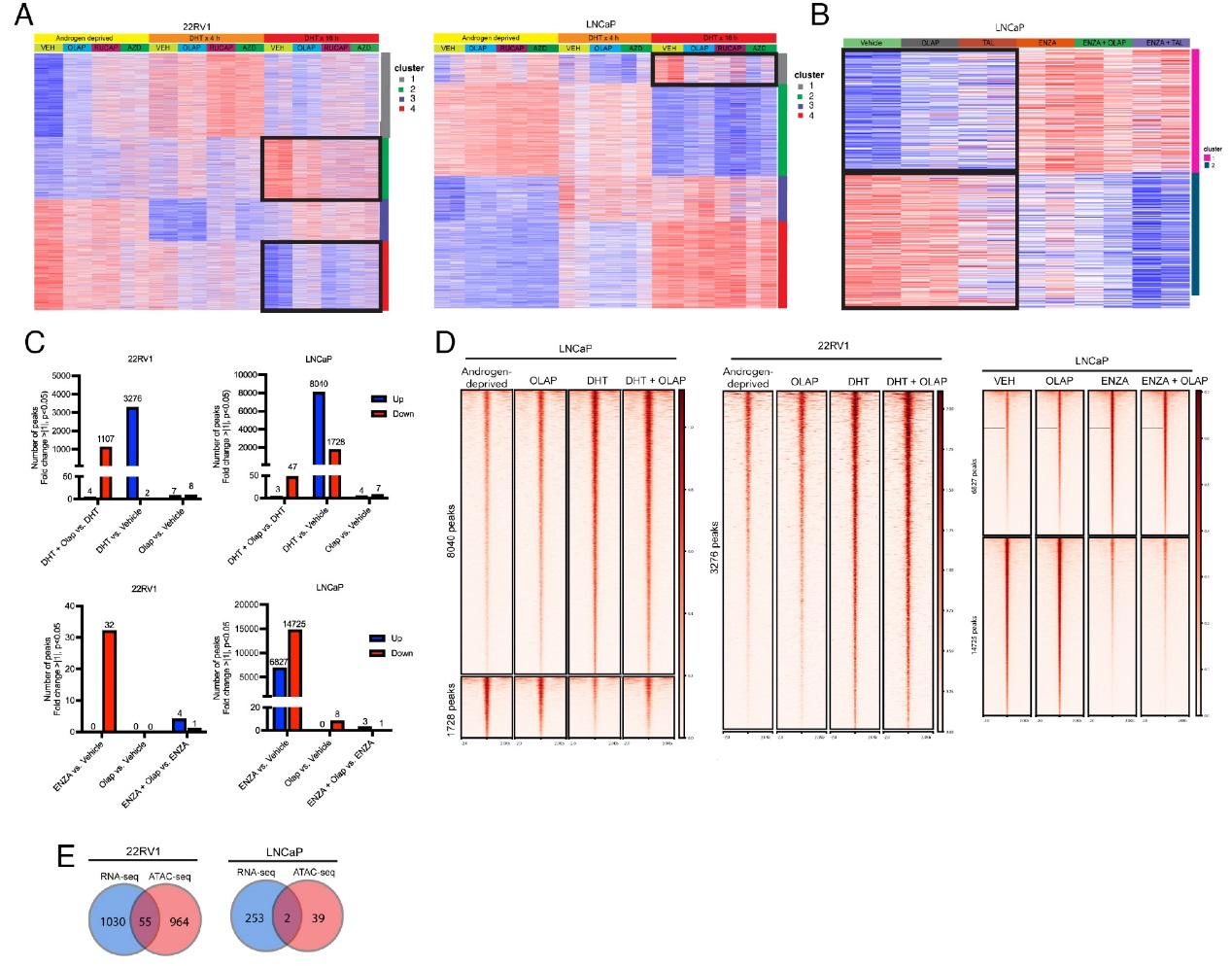
图5,抑制细胞周期进展会削弱对PARP抑制剂的治疗反应。
5. 全文总结
本研究dian覆了“AR抑制剂通过抑制DNA修复来协同PARP抑制剂"的传统理论,shou次揭示在HR正常前列腺癌中,细胞周期进展才是PARP抑制剂起效的关键前提。AR抑制剂会诱导G1期阻滞,从而削弱PARP抑制剂的疗效,这解释了二者联合疗法在HR正常患者中效果不佳的原因。该发现提示临床应避免对HR正常患者联合使用PARP抑制剂与细胞周期阻滞类药物(如ADT),同时支持继续探索该联合方案在HR缺陷患者中的应用。尽管研究主要基于细胞模型且深层机制有待阐明,但其结论为前列腺癌精准治疗提供了重要的理论转向与临床指导依据。
 欢迎来到
欢迎来到 联系人:张
联系人:张 地址:上海市长江南路180号长江软件园B区B637室
地址:上海市长江南路180号长江软件园B区B637室 邮箱:2844970554@qq.com
邮箱:2844970554@qq.com 传真:
传真: